Será que o segredo da juventude eterna se esconde no DNA de um rato pelado? Colocada nesses termos, a possibilidade parece absurda, mas o genoma do roedor desnudo em questão --o rato-toupeira-pelado, ou Heterocephalus glaber, como preferem os cientistas-- talvez traga pistas importantes sobre como mamíferos como eles e nós envelhecemos e lidamos com o câncer.
Isso porque essa criatura sui generis, cujo DNA decifrado está sendo apresentado na edição de hoje da revista científica "Nature", é o Matusalém dos roedores.
Arte
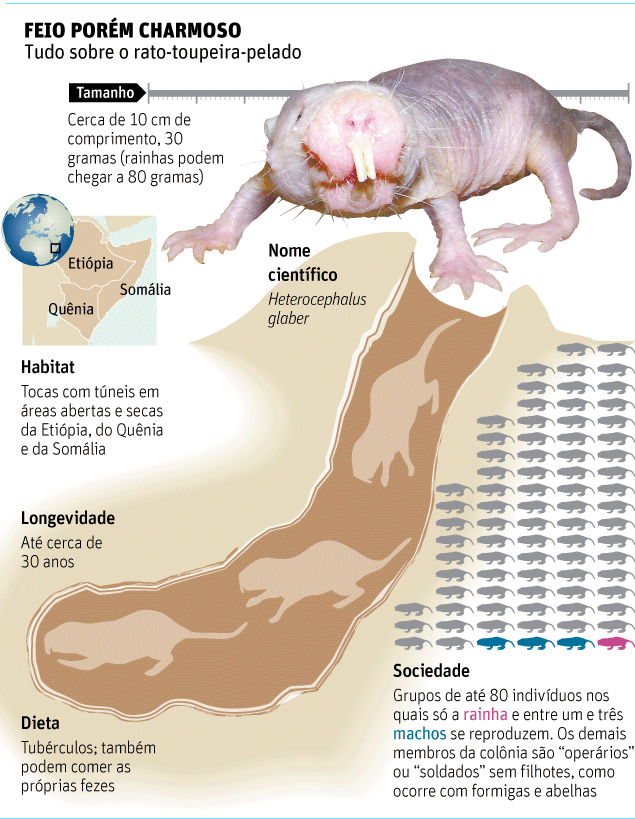
Enquanto ratos mais vagabundos, como os que povoam esgotos e biotérios de universidades, têm expectativa de vida de uns dois anos, o H. glaber, nativo da savana da África Oriental, pode até chegar à casa dos 30.
À PROVA DE CÂNCER
O bicho aparentemente consegue isso sendo extremamente refratário a tumores, por exemplo. Em laboratório, os pesquisadores têm até dificuldade de induzir a formação de cânceres na espécie.
Também é quase imperceptível o processo de envelhecimento das criaturas. A mortalidade não aumenta com a idade (menos quando se chega perto da longevidade natural do bicho, claro), e a fecundidade também é alta durante a vida toda.
Alta, quer dizer, para os poucos ratos-toupeiras-pelados que conseguem se reproduzir.
Ocorre que o bicho é o paralelo mais próximo com os insetos sociais (como abelhas e formigas) no mundo dos mamíferos. Isso significa que, como as abelhas, os ratos têm "rainhas" --e elas são as únicas fêmeas do grupo que se tornam mães.
Grandalhonas --de novo, assim como ocorre com as abelhas-rainhas--, as fêmeas reprodutoras têm características hormonais especiais, suprimindo o desenvolvimento das companheiras.
A rainha se une a um pequeno número de machos, enquanto o resto da colônia é estéril, adotando funções de operários (abrindo os túneis onde os bichos vivem) ou soldados (defendendo o grupo de invasores).
SEGREDO NAS PONTAS
Essa lista de características únicas pode ser vista por um novo prisma graças ao genoma recém-soletrado, trabalho que foi coordenado por Vadim Gladyshev, da Escola Médica de Harvard (EUA).
Para começar, os pesquisadores verificaram que, entre os genes do bicho que os diferenciam dos demais mamíferos, estão os que coordenam a estrutura dos telômeros --as pontinhas dos cromossomos, os quais abrigam o material genético. Os telômeros estão justamente ligados à divisão e ao envelhecimento das células.
A tendência é que, conforme as células se dividem e envelhecem, os telômeros encurtem --e isso leva a uma série de problemas bioquímicos. Os bichos, pelo jeito, acharam um modo de contornar esse fenômeno comum.
Os dados de DNA também indicam que a criatura é mais eficiente na hora de fazer uma faxina nas proteínas do organismo que sofreram danos. E também mantém em funcionamento pleno as mitocôndrias, usinas de energia das células, durante todo o seu período de vida.
Finalmente, os pesquisadores liderados por Gladyshev também acharam genes que ajudam o bicho a sobreviver em condições de baixo teor de oxigênio. Agora, o desafio é aplicar os dados em estudos sobre doenças que afetam seres humanos.
TEÓRICO PREVIU BICHO
Quem diz que a teoria da evolução é incapaz de fazer previsões sobre uma forma de vida que ainda está para ser descoberta deveria ser apresentado aos ratos-toupeiras-pelados.
Isso porque, nos anos 1970, o zoólogo americano Richard Alexander sugeriu que hábitos sociais como os de abelhas e formigas poderiam muito bem evoluir entre mamíferos, se as condições fossem adequadas.
Para que isso ocorresse, ele afirmou que a espécie em questão teria de ser um roedor, vivendo debaixo da terra nos trópicos africanos, em ninhos facilmente defensáveis e com fonte de comida abundante na forma de grandes tubérculos.
Na época, os ratos eram conhecidos, mas nada se sabia sobre seus hábitos. Alexander estava certo.